

The Bai Chen Research Group Publishes Paper in the International Journal of Molecular Sciences
Traditional drug design requires extensive research time and development costs. The burgeoning development of computational methods, including computational biology, computer-aided drug design, and artificial intelligence, has the potential to accelerate drug discovery efficiency by minimizing time and financial costs. In recent years, computational methods have been widely used to enhance the efficacy and effectiveness of drug discovery and pipelines, leading to the approval of numerous new drugs. The White Morning Research Group published a review article in the International Journal of Molecular Sciences, focusing on the application of computational methods in assisting drug target identification, lead compound discovery, and optimization. The article also discusses some challenges in using these methods for drug design. Additionally, the paper proposes a method to integrate various computational techniques into the discovery and design of new drugs.
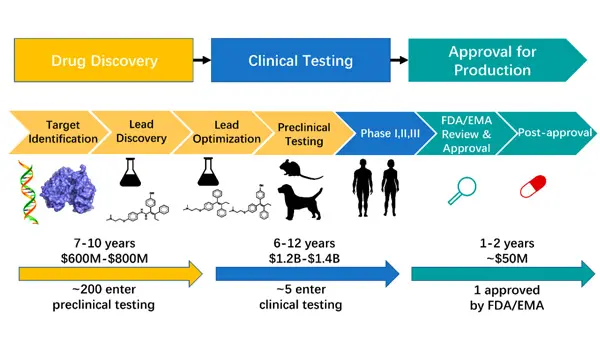
In the past forty years, details in drug development have undergone significant improvements. Today, the complete process of drug research includes drug discovery, clinical trials, and approval for production. The drug discovery process involves drug target identification, lead compound discovery, lead compound optimization, and preclinical trials. This process typically takes 7 to 10 years and costs between 600 million to 800 million dollars. About 200 compounds enter preclinical testing, with approximately 5 compounds proceeding to clinical testing, which consists of three phases: Phase I, Phase II, and Phase III clinical trials. This is a lengthy and costly process, taking 6 to 12 years and billions of dollars. Compounds that pass clinical trials enter the approval process for production. Compounds approved by FDA/EMA can be commercialized in the market. This process takes 1 to 2 years and costs approximately 50 million dollars.
Today, computational and deep learning methods play an increasingly important role in drug discovery. The rapid development of computational methods and algorithms has shortened the time and cost required to find candidate drugs. This paper systematically introduces the application of computational biology in drug design, computer-aided drug design, AI-driven de novo drug molecule design, and the technical approaches of AI tools in drug design.
This paper was co-authored by Dr. Zhang Yue, together with Dr. Luo Mengqi and research assistant Wu Peng. Additionally, Dr. Wu Song, the director of Shenzhen University-affiliated South China Hospital, provided assistance during the writing and publication process. Professor Li Zongyi and Professor Bai Chen are the corresponding authors of this paper.
分享:



Hot News
-
2022-11-05
The Bai Chen Research Group Publishes Paper in the International Journal of Molecular Sciences
- 2022-12-20
- 2022-12-26
- 2026-02-25